The main focus on the group concerns hierarchical self-assembly, and how such processes can be modelled. The first project with this theme was “Creating a framework for modelling the hierarchical self-assembly of anisotropic building blocks.” The two years of the project enabled the establishment of the research group, which is now itself growing hierarchically. The project was financed by UEFISCDI (project number PN-II-RU-TE-2014-4-1176), and ran from October 2015 to September 2017.
Objectives:
- creating a framework for modelling anisotropic building blocks across length scales, using advanced coarse-graining techniques
- studying the energy landscapes of selected hierarchically self-assembling systems
Abstract:
Hierarchical self-assembly is one of the most promising tools in nanotechnology. In biological systems, such processes have been already perfected during evolution, and involve successive formation of building blocks from smaller units, which in turn self-assemble into larger structures in a hierarchical fashion (e.g. virus capsid proteins, keratin filaments, amyloid fibrils etc.). Presently, computational modelling of assembly processes on the nanoscale is possible only with coarse-grained methods, chosen appropriately for the size of the system and the particular problem. However, hierarchical self-assembly often happens across multiple scales, and each layer of the process has to be modelled differently: molecular mechanics force fields for protein folding, united atom force fields for oligomerization, shape-based coarse-grained models (e.g. bead models) for successive assembly of protein oligomers. To this date, no framework exists supporting modelling such processes across length scales.
The aim of the project is to use the experience of the PI with modelling anisotropic building blocks using the rigid body framework, and extend the method to bridge the gap between the different length scales. The method will involve a hierarchical calculation of building block parameters, obtained from extensively studying the energy landscape of their components. This approach will be scale-independent, and will allow bottom-up design of novel complex structures on the nanoscale.
Project results:
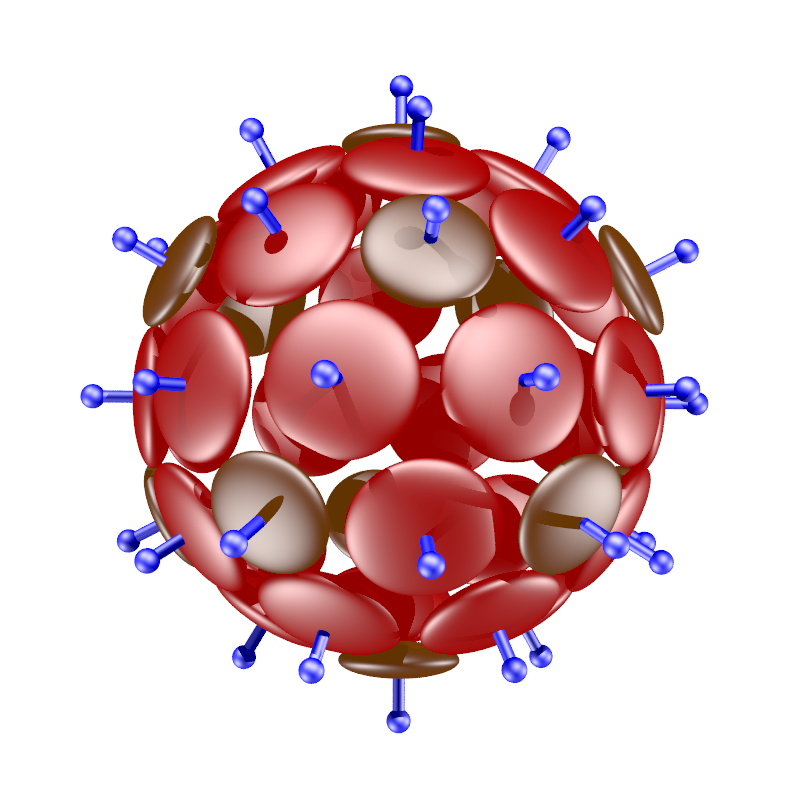
During the project, we created new methods and algorithms for determining coarse-grained parameters representing the shape of a building block modelled with atomistic potentials. Another priority was to determine the necessary conditions for selected building block shapes that determine their hierarchical self-assembling properties. We have created minimal physical models enabling hierarchical self-assembly into exotic structures such as Goldberg polyhedra, stacked rings, dodecahedral cages etc.
Summary of scientific activities between 01.10.2015 – 01.12.2016:
– development of hierarchical coarse-graining methods
– studying the aggregation of CCMV capsid proteins
– devising new design principles for hierarchical self-assembly by using macroions
A detailed version of the progress report can be downloaded from here.
Summary of scientific activities between 01.12.2016-30.09.2017:
– adaptation of the developed hierarchical coarse-graining methods for capsid proteins
– studying the dynamics of CCMV protein dimers, microsecond MD simulations
– designing addressable building blocks capable of hierarchical self-assembly
– creating a minimalistic model for hierarchical self-assembly of polyhedral shells of exotic symmetries
– coarse-grained modelling of spherical ‘Blackberry’ structures
Computing resources:
Our computations are done on a small compute cluster by Supermicro, with three performant Nvidia Tesla K40M GPUs, and 44 CPU cores. Details of the architecture can be found here.
Contact:
The project was implemented in the R&D department of Provitam Foundation. Address: 16 Muncitorilor street, Sfantu Gheorghe, Romania.
Media
This video illustrates how kinetically trapped structures form in a system capable of forming a Kagome lattice (triblock Janus particles). This is a constant energy MD run adapted for ellipsoidal rigid bodies. Videos about large-scale rearrangements of self-assembling coarse-grained systems can be found on my Youtube channel.
The second project of the group built on the first one, and had the title “Coordination chemistry on the nanoscale: Computational design of supramolecular building blocks capable of highly specific, orientation-dependent interactions.” That project was financed by CNCS-UEFISCDI as well (project number PN-III-P1-1.1-TE-2016-1279), and ran from May 2018 to April 2020.
Abstract
With the advent of modern synthetic methods of nano- and mesoscale building blocks, it is now possible to encode complex self-assembling behaviour in relatively simple particles. However, the parameter space available for experimentalists is huge: one has to tune the building block anisotropy, interaction anisotropies, range, type etc. Computational methods can add valuable insight into the rational design of such building blocks. Coarse-grained modelling of anisotropic interactions can guide experiments into regions of the parameter space relevant to the desired target self-assembled structure.
The project aim was to establish a new field for the self-assembly of nanoscale building blocks, through applying concepts from coordination chemistry into designs of nanoparticles, which will become capable of highly specific coordination to other nanoparticles. Although the concept of ‘colloidal molecules’ exists, experimental realizations are still in their infancy. In order to understand the behaviour of such building blocks, and to create the simplest possible models for them, we will be using and further developing state-of-the-art methods in energy landscape theory (global optimization within the rigid body framework, discrete path sampling and rigid body MD).
In the first stage, we explored the minimal physics required for assembly of hollow cages formed by nanoscale analogues of MnL2n-type Goldberg polyhedra, which are experimentally realized by square planar coordinated transition metal complexes with nonlinear bidentate ligands. We found that cages with the same symmetry can be obtained using a combination of excluded volume and Coulombic interactions, and investigated the dynamics of hollow shell formation and transition between competing structures. The second stage involved the design of novel anisotropic nanoparticles capable of tetrahedral, planar and linear coordination, giving rise to mesoscale structures analogous to hydrocarbons.
Summary of research activities
- improving Monte-Carlo exploration for basin-hopping global optimization of colloidal particles
- optimizing the evaluation of rigid-body potentials on GPUs
- implementing the developed potentials into OPTIM
- creating an algorithm for seeded global optimization
- global optimization of MnL2n-type shells
- disconnectivity graphs for clusters of 24 and 30 central particles forming shells of different symmetries
- preliminary MD confirmation of self-assembly of shells with exotic symmetries
- long MD runs for macroions in explicit water
- long MD runs for ss-CCMV capsid protein dimers and pentamers of dimers
A detailed progress report can be downloaded from here (stage 1) and here (stage 2).